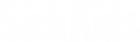|
BACKGROUND
According to recent estimates, splice site defects account for 5% to 10% of pathogenic mutations causing Mendelian diseases. The prevalence is even higher for neuromuscular disorders (NMDs) due to the unusually large size and the multi-exonic structure of genes encoding muscle structural proteins, further highlighting the importance of these mutations in NMDs. Therapeutic genome editing can be exploited to correct disease-causing mutations. However, in previous studies, correction of splice site mutations have only been accomplished via the homology-directed repair (HDR) pathway, which is extremely inefficient in post-mitotic tissues such as skeletal muscles hampering its therapeutic utility in NMDs.
DESCRIPTION OF THE INVENTION
SickKids researchers have demonstrated a novel strategy to correct a pathogenic splice site mutation by harnessing the non-homologous end-joining (NHEJ) repair pathway.
As a proof-of-principle, they focused on merosin-deficient 34 congenital muscular dystrophy type 1A (MDC1A), which is characterized by severe muscle wasting and paralysis. Specifically, they corrected a splice site mutation in the Lama2 gene, which causes exclusion of exon 2 and truncation of Lama2 protein in dy2J/dy2J mouse model of MDC1A. Using adeno-associated viral vector serotype 9 (AAV9) to deliver S. aureus Cas9 and two guide RNAs, they simultaneously excised the intronic region containing the mutation and created a functional splice donor site through NHEJ.
This strategy led to successful inclusion of exon 2 in the Lama2 transcript and restoration of full-length Laminin-α2 protein. Importantly, the treated dy2J/dy2J mice display significant improvement in muscle histopathology, strength and function without any signs of paralysis.
COMMERCIAL APPLICATIONS & ADVANTAGES
This discovery demonstrates an innovative approach that is independent of the HDR pathway to correct a non-coding mutation by modifying an intronic region to create a functional splice donor site. Given that a significant proportion of MDC1A individuals are affected by splice site mutations, this strategy carries a therapeutic potential for numerous patients. Furthermore, it highlights a far-reaching therapeutic potential and translatability of this strategy for diseases caused by splice site mutations.
This discovery has tremendous market potential since it would benefit the majority of patients with donor splice site mutations which accounts for approximately 5%–10% of all reported human mutations.
DEVELOPMENTAL STAGE
- Pre-clinical in vitro data available
- Pre-clinical in vivo mouse data available
PUBLICATIONS
- Nature Medicine 23, 984–989 (August 2017)
PATENT STATUS
- US Prov. Appl. # 62/530,703 filed Oct 4, 2016
- US Pat. Appl. # 16,629,883 filed Jan 9, 2020
- CA Pat. # 3,069,338 filed Jan 9, 2020
- EP Pat. Appl. # 17917339.8 filed Jan 9, 2020
|
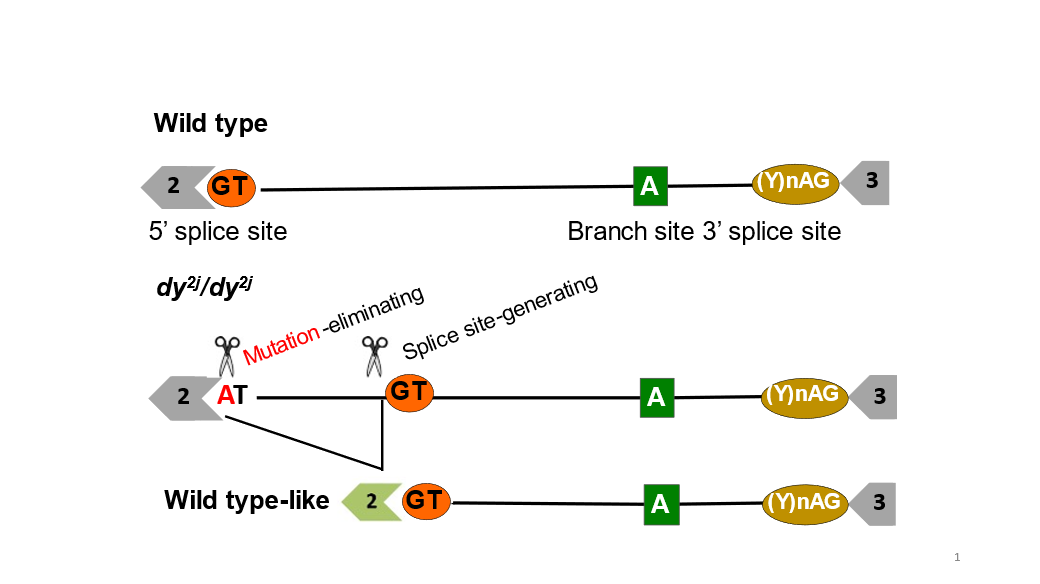 Figure 1. A Homology Recombination independent strategy to repair normal splicing.
Figure 1. A Homology Recombination independent strategy to repair normal splicing.
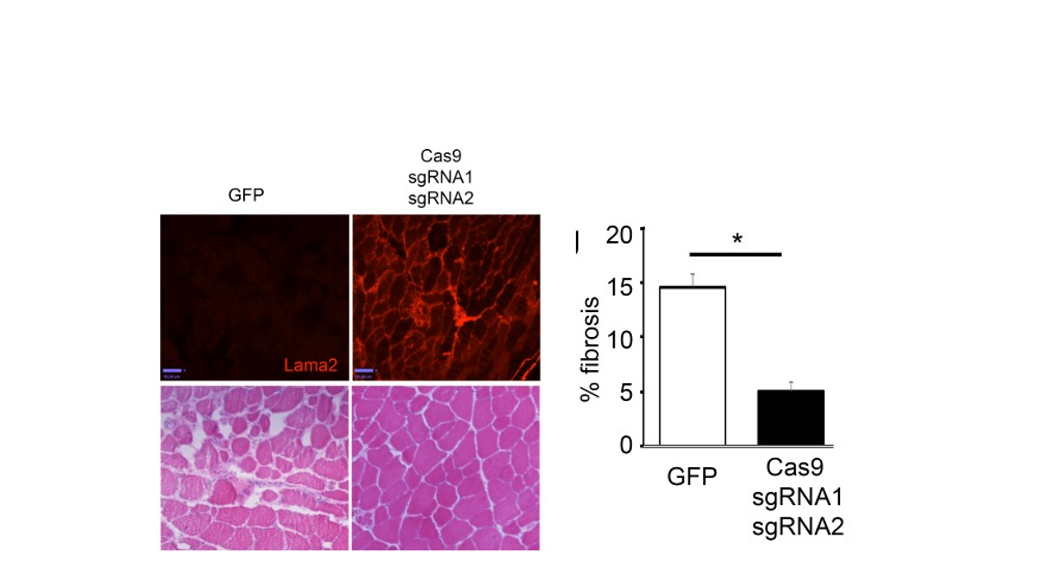 Figure 2. Restoration of full-length Lama2 protein improves muscle histopathology.
Figure 2. Restoration of full-length Lama2 protein improves muscle histopathology.
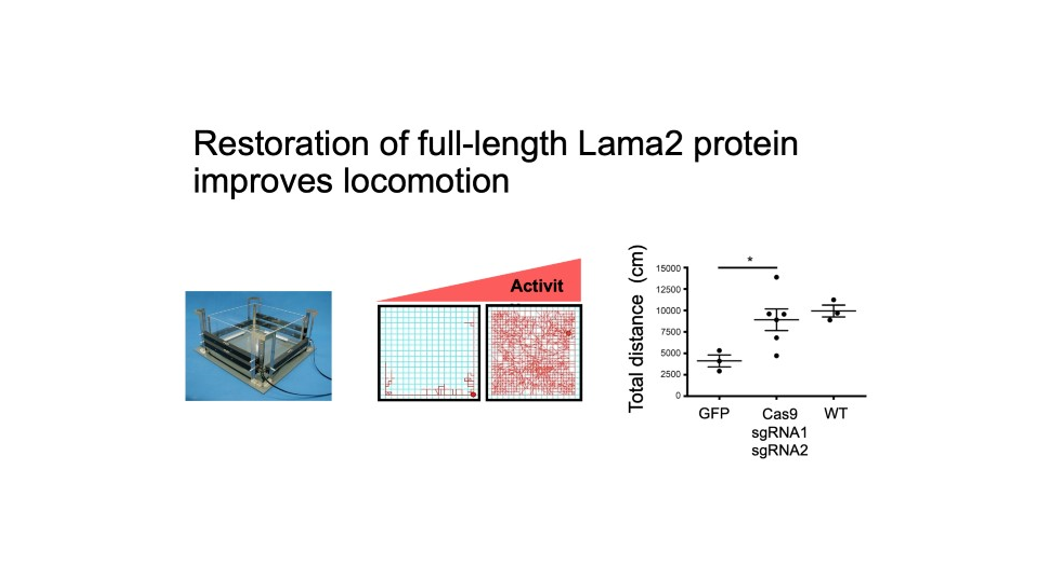 Figure 3. Restoration of full length Lama2 improves locomotion.
Figure 3. Restoration of full length Lama2 improves locomotion.
|